Quantum computing represents a computational paradigm that leverages some unique properties of quantum systems to perform calculations that are infeasible for classical computers. Various computational tasks, such as electronic structure simulation of molecules which is crucial for understanding the atomistic properties of materials, can be executed more efficiently on quantum computers. Although we do not yet have a quantum computer capable of performing useful computations faster than classical computers, the advent of such devices will enable calculations at speeds orders of magnitude greater than those achievable with our most advanced supercomputers. For further information on quantum algorithms that facilitate these computations, please refer to the following references in my Publications page: 15, 18, 19, 20, 22, 23, 24, 25, 26, 27, 28, 29, 30.
Point processes are statistical models that can be used to simulate random point patterns. Algorithms to generate samples from structured point processes can be applied to machine learning tasks such as clustering and feature selection. By encoding similarity or correlation structures in graphs or matrices, such algorithms preferentially sample more informative subsets, improving performance in classical learning algorithms. These quantum-inspired methods show potential for enhancing data-driven models, especially where identifying relevant features or patterns is computationally intensive. For further information on point processes in machine learning, please refer to the following references in my Publications page: 18.
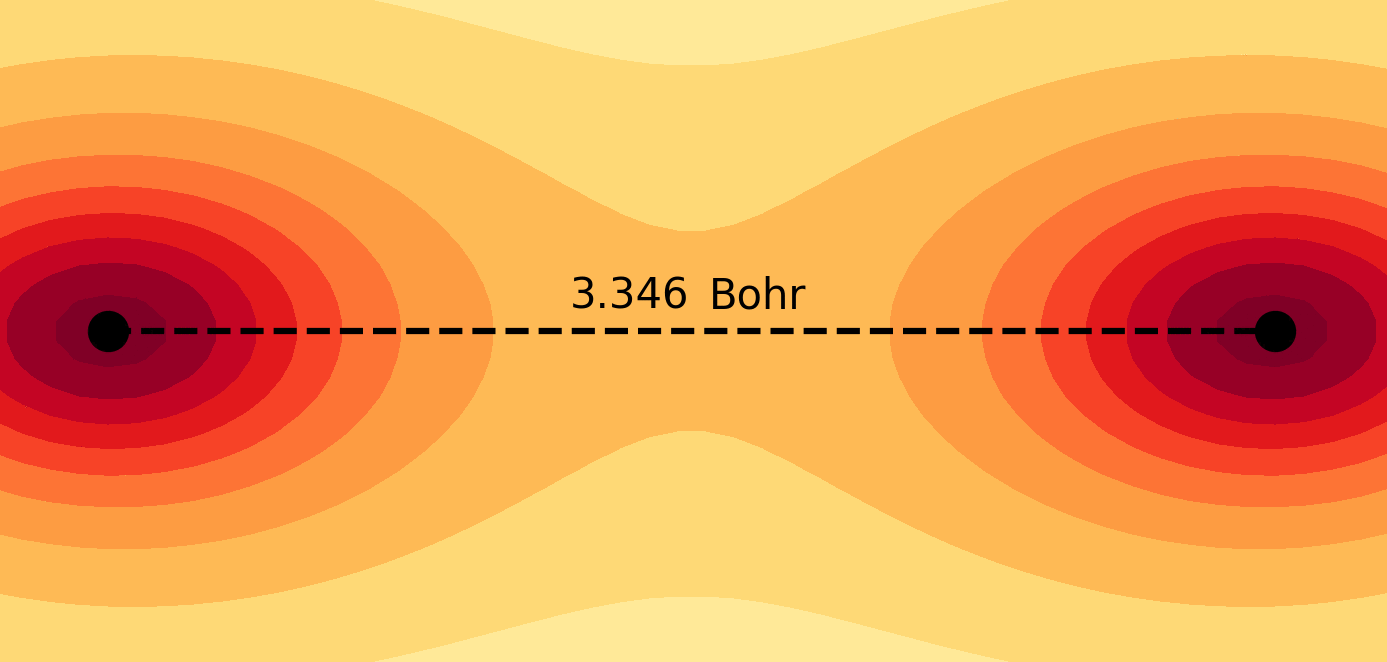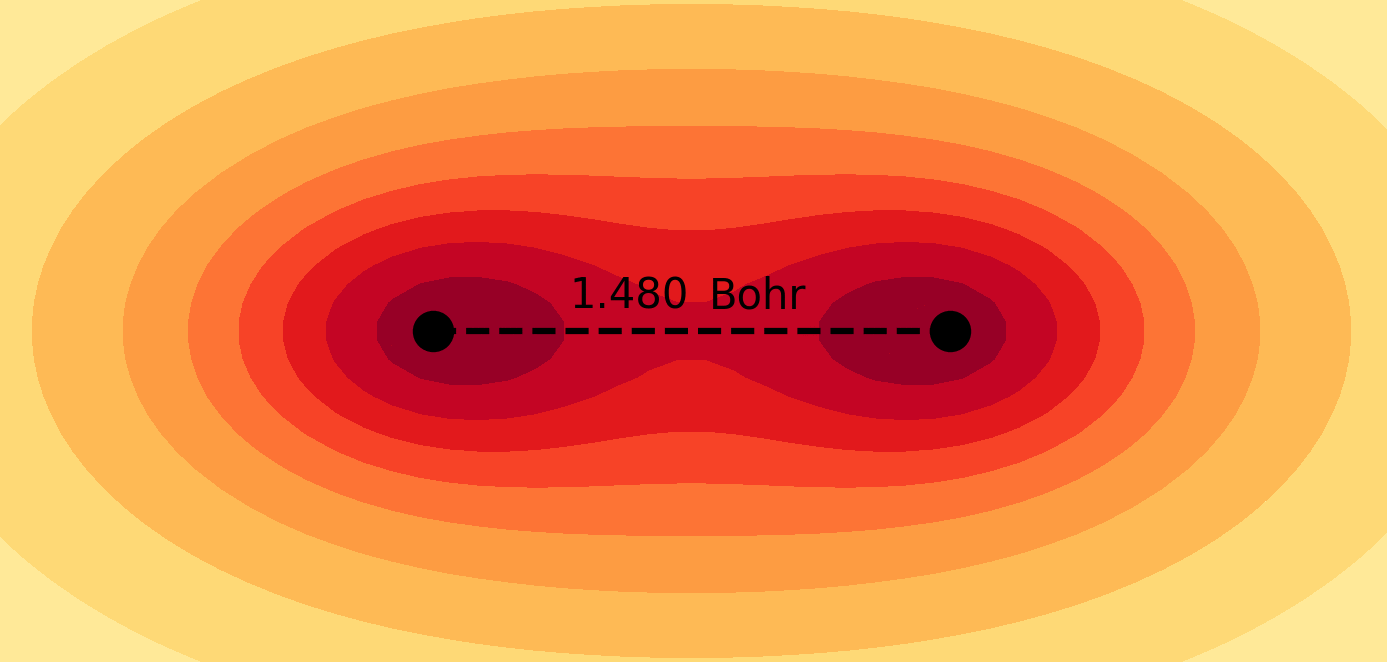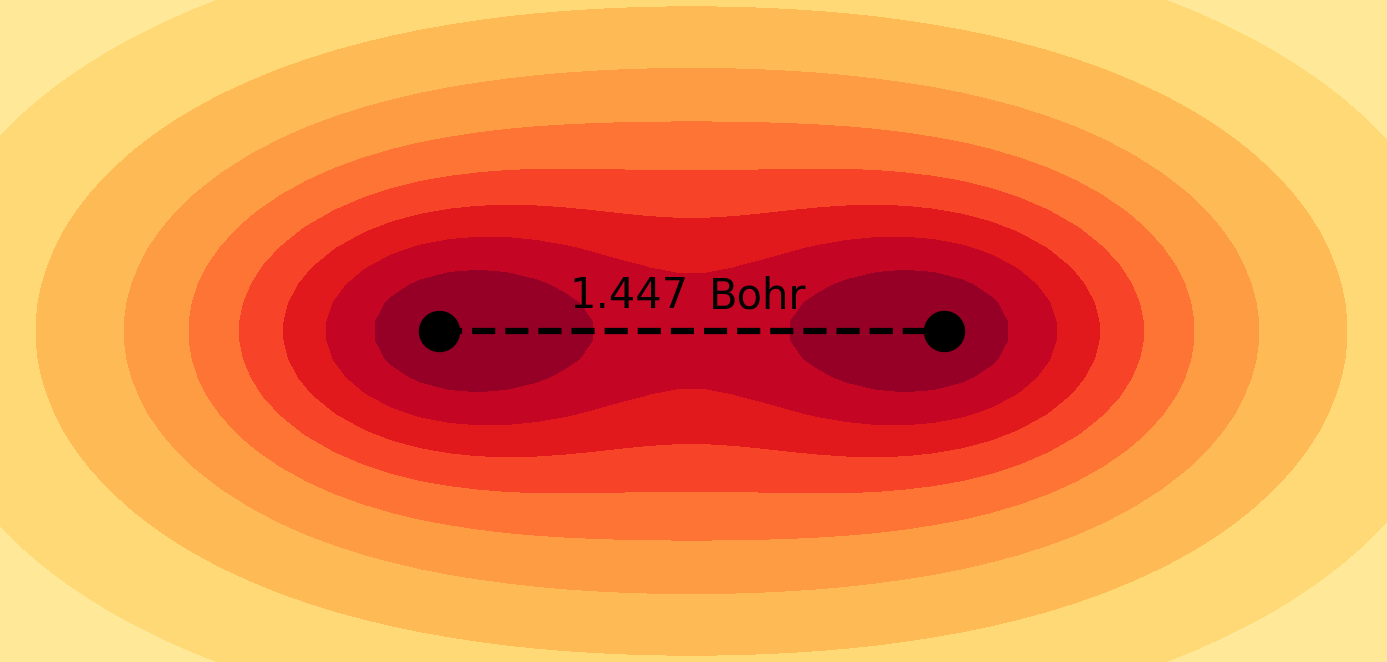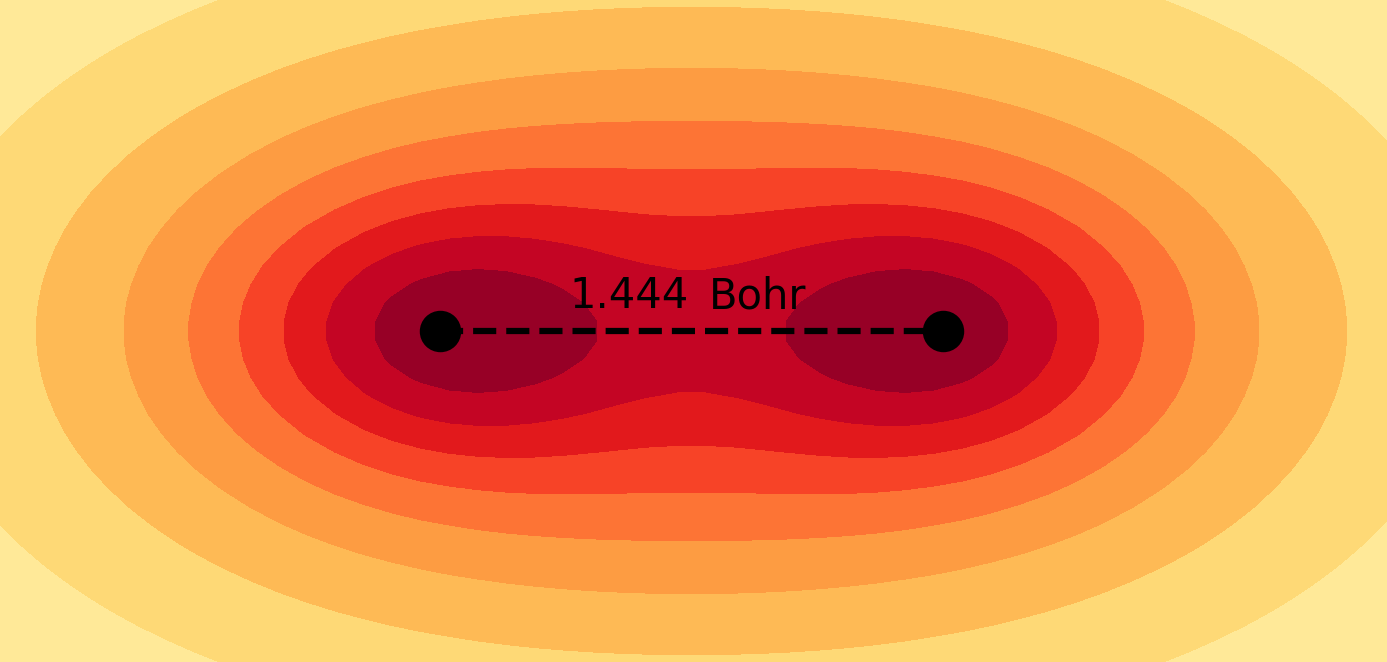
Molecules and materials can be simulated by constructing them from their subatomic components using the principles of quantum mechanics. These simulations provide valuable information about the electronic structure and chemical properties of materials that are typically difficult to obtain through experimental methods. Quantum chemical simulations are routinely performed to investigate the mechanisms of chemical reactions and explore the atomistic structure of nanosystems, which are essential for understanding fundamental chemical processes in both nature and technology. For more details on the application of molecular quantum simulations, please refer to the following references in my Publications page: 4, 6, 9, 10, 11, 12, 13, 14, 17, 21.
Molecules are not static entities with fixed structures; they exhibit internal dynamics, such as vibrations, and move through their environment, continuously interacting with other molecules. The behavior of molecules during chemical processes, like the diffusion of a drug through a cell membrane, can be simulated at the atomic level. These simulations generally treat atomic motion classically, while the interactions between atoms can be described using either quantum mechanics or classical force fields. Molecular dynamics simulations offer detailed atomistic insights, enhancing our understanding of chemical processes. For more information about these simulations, please refer to the following reference in my Publications page: 3, 7, 9, 11, 16.
soranjh.github.io